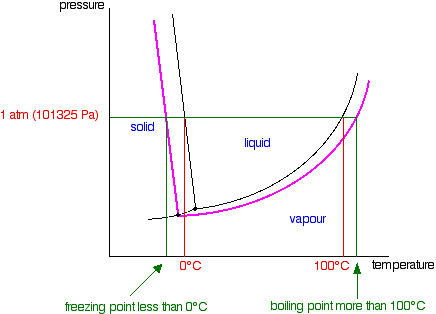
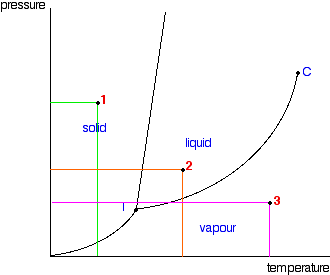
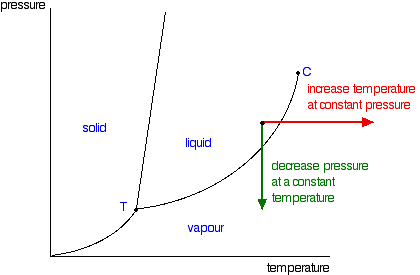
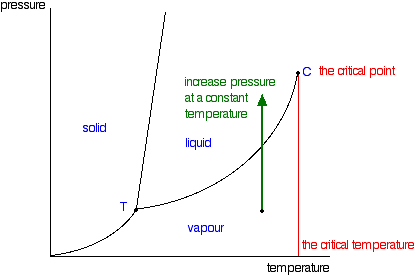
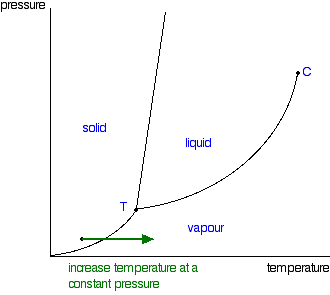
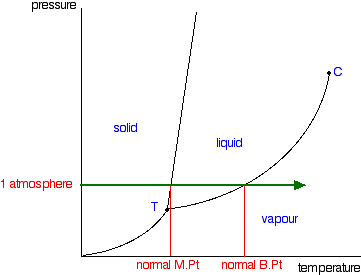
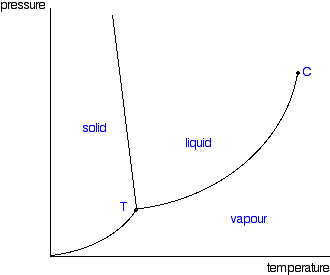
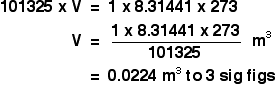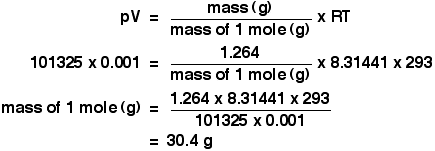It is a type of cell division that occurs only in the reproductive cells and hence it is known as germ cell division. The daughter cells resulting from meiosis will carry half the number of chromosomes and half the amount of genetic material compared to the parent cell. Hence, meiosis is also known as reductional division.

fig. 17.5 - Stages of Meiosis
Meiosis occurs during the formation of gametes (spermatozoa and ova) in animals and spores in higher plants. It is mainly meant for bringing about a decrease in the chromosome number from the diploid (2n) condition to the haploid (n) condition.
The most characteristic feature in meiosis is that nucleus divides twice successively. Hence, meiosis is conventionally divided into first meiotic division (or meiosis - I) and second meiotic division (or meiosis - II).
In both the meiotic divisions, karyokinesis can be distinguished into four stages - prophase, metaphase, anaphase and telophase.
First Meiotic Division
The first meiotic division or meiosisI is commonly described as reductional division since at the end of this division the two resulting daughter cells will have half the number of chromosomes as that of the parent cell.
Interphase
It is the preparatory phase. Cell organells replicate and size of the cell increases. DNA molecule undergoes replication. Each chromosome exists as a pair of chromatids joined together by a centromere.

fig. 17.6 Stages of Meiosis-I
Prophase I
It is the phase of longest duration and involves a series of significant changes in the chromosomes. These changes are often described in five substages namely leptontene, zygotene, pachytene, diplotene and diakinesis.
Leptotene
- : Chromosomes shorten and become visible as single structures. In some cases they have a beaded appearance showing densely staining material called chromomeres alternating with nonstaining regions.
Zygotene
- : Paternal and maternal chromosomes come together and pair up. This pairing of homologous chromosmes is called synapsis. The paired chromosomes are described as bivalents. The bivalents shorten and thicken (spiralisation).
Pachytene
- : Each chromosome splits into two chromatids and thus each pair will have four chromatids two paternal and two maternal. They are now called tetrads. The non-sister chromatids of the paternal and maternal chromosomes overlap each other. They appear to be joined at several regions along their length. These points are called Chiasmata. Each chiasma is the site of an exchange of genetic material between the two chromatids. It occurs due to breakage and reunion between the two non-sister chromatids. This process is called genetic recombination.
Diplotene
- : The synaptic forces holding the two chromosomes in the pair come to an end. The chromosomes start separating. This separation is called as disjunction.
Diakinesis
- : Separation of the chromosomes is now complete with paternal and maternal chromosomes having exchanged portions of chromatids. The chromosomes condense again. The chiasmata disappear by sliding towards the tips of the chromatids. This process is called terminalisation.
By the time these changes are completed in the chromosomes, the nuclear membrane and nucleolus disappear. Asters and spindle fibres make their appearance.

fig. 17.7 - Behaviour of Chromosomes during Meiosis-I
Metaphase-I
In this change of a very brief duration, the chromosomes move towards the equator of the cell and come to lie in two parallel metaphase plates. These two parallel plates are formed by one set each of the homologous chromosomes. Each homologous chromosome has two kinetochores, one for each of its two chromatids.
Anaphase-I
There is no splitting of the centromere. As a result homologous chromosomes of each pair rather than the chromatids of a chromosome separate and move to the opposite poles. As a result, half the number of chromosomes that appear in the early prophase, move to each opposite pole. It is here that an actual reduction in the chromosome number (from (2n) to (n)) occurs. However, each chromosome found at the poles consists of two chromatids.
This is in contrast to the single stranded chromosomes in the anaphase of mitosis.
Telophase-I
The chromosomes at each pole uncoil and elongate to form the chromatin. A nucleolus reappears at each pole. Spindle fibres and asters disappear and centrioles split. A nuclear membrane is formed at each pole resulting in the formation of two daughter nuclei.
Cytokinesis - I
Simultaneously with the formation of two daughter nuclei, a cleavage furrow appears in the middle of the cell. The furrows gradually deepen and divide the cell into two daughter cells. Each of the resulting daughter cell prepares itself to undergo the second meiotic division.
Interkinesis
There is no interphase preceding second meiotic division. There is a brief intervening period called interkinesis. During this period there may be synthesis of some reserve food and proteins. However, there is no replication of DNA prior to meiosis II.
Second Meiotic Division
The second meiotic division or meiosis II almost always follows the first meiotic division. This division is primarily meant for separating the two chromatids of each chromosomes. Meiosis-II also has a Karyokinesis and a cytokinesis.
Karyokinesis of meiosis-II (Karyokinesis-II) can be distinguished into four stages namely prophase-II, metaphase-II, anaphase-II and telophase-II.
Prophase-II
It is of a short duration compared to prophase-I. No significant changes take place in the chromosomes. Nuclear membrane and nucleolus disappear. Asters and spindle fibres are formed.
Metaphase-II
The chromosomes line up at the equator of the cell forming a single metaphase plate (as in mitosis).

fig. 17.8 - Stages of Meiosis-II
Anaphase-II
The centromere splits and two chromatids in each chromosome start moving away from each other. Finally, they reach the poles of the cell. Each pole now has haploid number of chromosomes and half the amount of DNA.
Telophase-II
Chromosomes at each pole uncoil and elongate to form chromatin. Nucleolus and nuclear membrane are formed surrounding each chromatin network. Asters and spindle fibres disappear and centrioles divide. Daughter nuclei are formed.
Cytokinesis-II
A cleavage furrow appears in the middle of the cell, deepens gradually and divides the cell into two.
Thus, at the end of second meiotic division, four daughter cells are formed. Each daughter cell has not only half the number of chromosomes but also half the amount of DNA, as that of the parent cell. Thus, the resulting cells are truly haploid cells.
Significance of Meiosis
Meiosis becomes significant for the following reasons.
- It brings about a reduction in the chromosome number from a diploid (2n) condition to a haploid (n) condition. Such a reduction becomes necessary for maintaining the chromosome number.
- It provides chance for the appearance of new gene combinations as a result of crossing over. This situation brings about variations.
- It is a division necessary for the formation of gametes in animals and spores in plants.
Both mitosis and meiosis essentially follow the same sequence in all living organisms, which is an evidence of the basic relationship between diverse groups of living organisms